Abstract
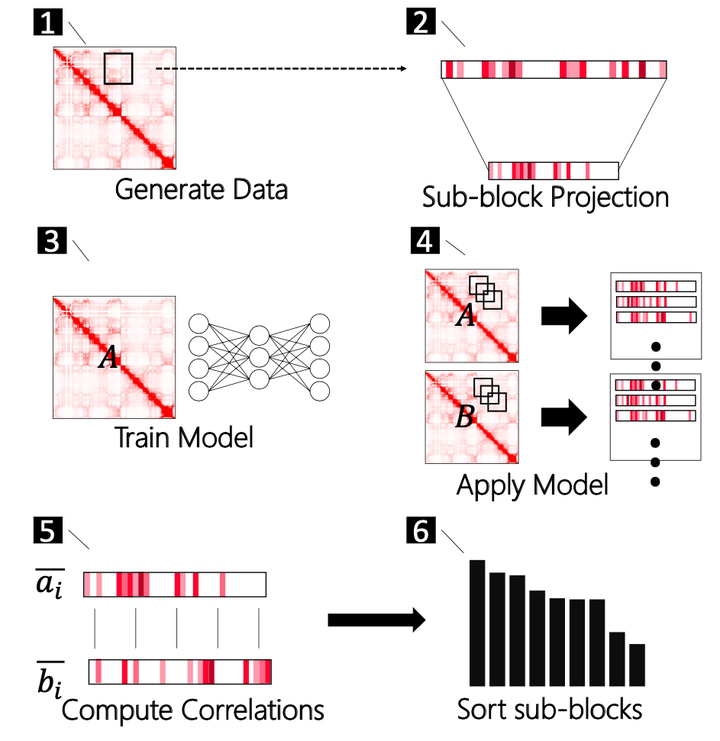
The development of high-throughput chromosome conformation capture techniques (Hi-C) has provided a wealth of data on the three-dimensional architecture of genomes. We can use this data to analyze the topological structure of genome and understand genomic interactions. However, the accurate approach to find conserved or specific genomic interactions in two or more Hi-C contact matrices is an open question. We introduce a convolutional autoencoder an unsupervised machine learning technique to produce a similarity function to compare areas between pairs of Hi-C matrices. Our model is trained on sub-blocks of the Hi-C matrix that are treated as high-dimensional vectors and that are transformed into lower dimensional vectors. We show that our autoencoder outperforms statistical methods such as principal components analysis (PCA) and root-mean-square error (RMSE) for finding genomic interactions which are specifi c to one of the matrices. This method is useful and accurate in finding genomic interactions specifi c to one genome which potentially result in changes in gene expression by comparing Hi-C matrices from two or more tissues or species.
Carlos Rojas
Assistant Professor
My research interests include bioinformatics, computing education, visualization, and machine learning.